弥漫性大 B 细胞淋巴瘤(Diffuse large B-cell lymphoma, DLBCL)是最常见的非霍奇金淋巴瘤(non-Hodgkin lymphoma, NHL)类型,约占美国新诊断的 B 细胞 NHL 病例的 22%。每年约有超过 18,000 人被诊断出患有 DLBCL。DLBCL 具有高度异质性的遗传异常,导致不同患者的临床表现、治疗反应和结果显著不同。以蒽环类药物(即环磷酰胺、长春碱、多柔比星和泼尼松(CHOP))为基础的化疗是治疗 DLBCL 的主要手段,经过化疗的半数患者可长期生存或治愈。在 CHOP(R-CHOP)化疗中加入利妥昔单抗(rituximab, R)可将长期生存率进一步提高约 20%。然而,其未能满足难治性和复发性患者的临床需求。
MicroRNAs(miRNAs)是一种内源性小非编码 RNA 分子(约 22 个核苷酸),可通过与靶转录物 3'-非翻译区(UTR)中的部分互补位点结合来抑制目的基因的表达。miRNA 已被公认可用于有效区分不同的 B 细胞淋巴瘤,并参与疾病的发展、进展和临床反应。其中,miRNA-17-92 簇在 DLBCL 中高度表达,与 MYC 的合作可以加速 B 细胞淋巴瘤的发生。得注意的是,miRNA-106a-363 簇作为 miRNA-17-92 家族的另一个成员,据报道也发挥潜在的致癌作用。之前的研究显示 miRNA-363-3p 与 DLBCL 中 R-CHOP 治疗失败显著关联。
2022 年 4 月 29 日,郑州大学附属肿瘤医院的研究人员在 Leukemia 杂志上发表了题为“MiRNA-363-3p/DUSP10/JNK 轴通过增强弥漫性大 B 细胞淋巴瘤的 DNA 损伤修复介导化疗耐药”的研究论文。该研究测定了 miRNA 和基因表达谱以揭示潜在的化学抗性机制和治疗方法,观察并验证了 miRNA-363-3p 的高表达与化学抗性之间的独立相关性。MiRNA-363-3p 通过异位表达和 CRISPR/Cas9 介导的 DLBCL 细胞系敲除,在体外和体内实验中被证明能减少多柔比星诱导的细胞凋亡和肿瘤萎缩。miRNA-363-3p/DUSP10/JNK 轴主要与同源重组(HR)和 DNA 修复途径的负调节有关。靶向 JNK 和聚(ADP-核糖)聚合酶 1 能显著抑制多柔比星诱导的 DSB 修复,增加多柔比星诱导的细胞凋亡和肿瘤萎缩,并改善肿瘤小鼠的生存。总之,miRNA-363-3p/DUSP10/JNK 轴是 DLBCL 中一种新的化疗耐药机制,可能被靶向治疗逆转。
对来自 47 名 DLBCL 患者的冷冻肿瘤样本进行了 miRNA 表达谱分析。miRNA-363-3p 的水平在耐药组中显著增加,也高于 naive 组、中胚层和中心细胞 B 细胞(图 1A)。包括基于 GEP 的 COO 分类和 IPI 评分的临床参数在内的多变量分析显示 miRNA-363-3p 与耐药性之间存在独立关联。没有发现关于 miRNA-363-3p 的其他旁系同源物的显著差异,这些类似物在前体 miRNA 分子的二级结构上表现出明显的差异。此外,具有高 miRNA-363-3p 水平的患者显示出不利的无进展生存(Progression-free survival, PFS)(图 1B)。
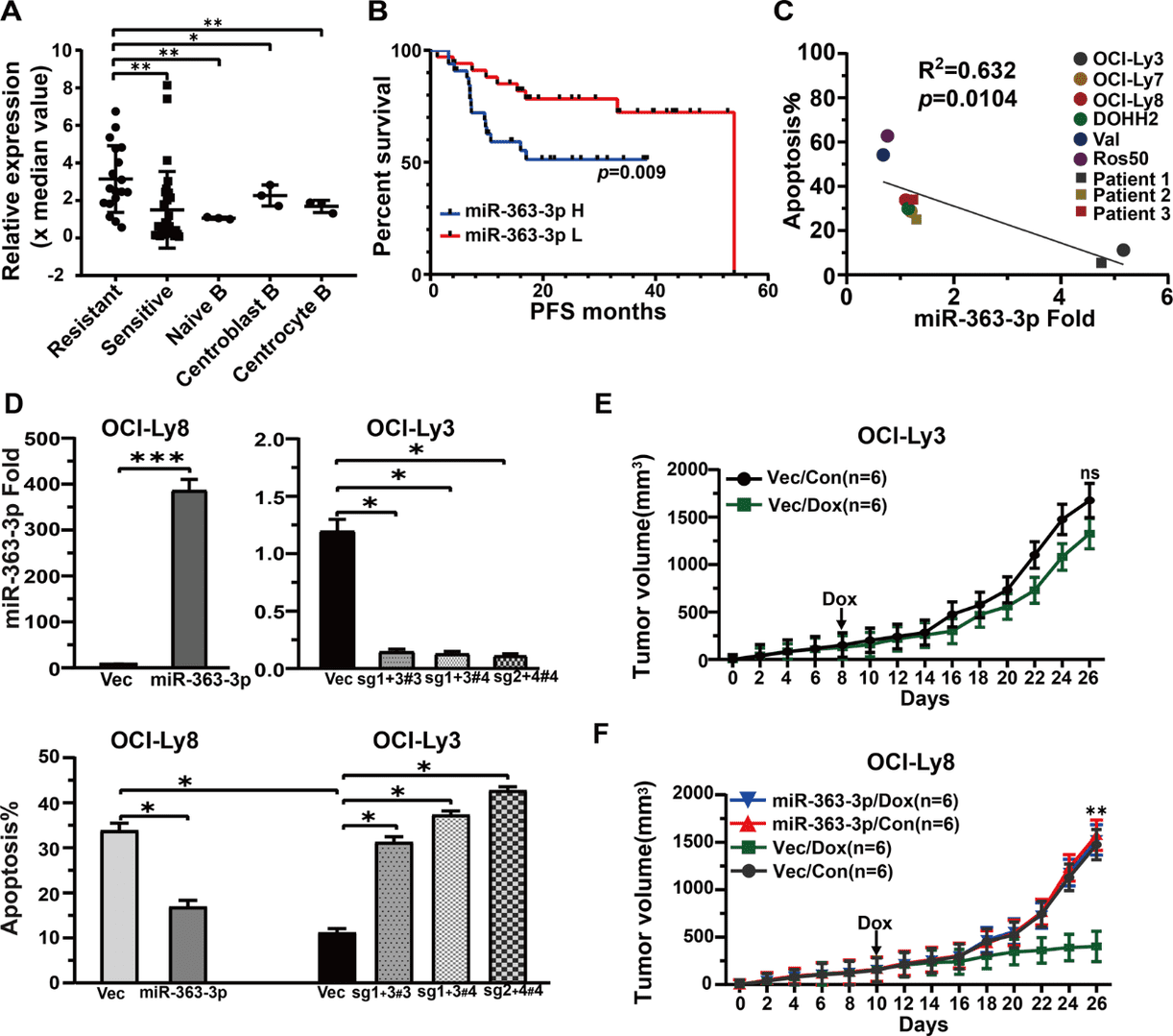 图1. miRNA-363-3p 的高表达与化疗耐药性之间存在显著关联。
图1. miRNA-363-3p 的高表达与化疗耐药性之间存在显著关联。
在 B 细胞淋巴瘤细胞系中也观察到 miRNA-363-3p 水平的高度变化。处理 48 h 后,6 个 DLBCL 细胞系和 3 个 DLBCL 患者肿瘤细胞中 miRNA-363-3p 的水平与多柔比星(25 ng/ml)诱导的细胞凋亡呈负相关(图 1C)。在 miRNA-363-3p 异位的 OCI-Ly8 细胞系中观察到多柔比星诱导的细胞凋亡显著减少,而在 CRISPR/Cas9 介导的 miRNA-363-3p 敲除的 OCI-Ly3 细胞系中检测到细胞凋亡显著增加(图 1D)。体内实验证实了 miRNA-363-3p 表达与多柔比星耐药性之间的关系,观察到 miRNA-363-3p 敲除的 OCI-Ly3 细胞系具有低的肿瘤发生效率。事实上,细胞活力测定表明敲除 miRNA-363-3p 显著抑制细胞增殖。荷瘤小鼠被分为对照组和实验组(每组 n = 6)。与携带 OCI-Ly3 载体的小鼠相比,多柔比星在携带 OCI-Ly8 载体的小鼠中诱发了明显的肿瘤缩小,并且多柔比星对 OCI-Ly8 的疗效因 miRNA-363-3p 的异位表达而显著降低(图 1E、1F)。
围绕 CpG 岛的 1.258 kb 序列被预测为候选的 miRNA-363-3p 启动子,其功能通过与阴性对照相比明显增强荧光素酶活性而得到验证(图 2A)。生物信息学分析揭示了 CpG 岛中 MYC 结合的四个 E-box 元件。通过染色质免疫沉淀加 PCR 测定法将 E-box-2 确定为 MYC 的精确结合位点。值得注意的是,OCI-Ly3 中 MYC 的结合水平高于 OCI-Ly8中(图 2B)。通过亚硫酸氢盐测序 PCR 进行的位点特异性 DNA 甲基化分析发现,OCI-Ly3 中 E-box-2 周围的 CpG 甲基化程度低于 OCI-Ly8(57% 对 85%,p < 0.05)(图 2C)。用 DNA 甲基转移酶抑制剂地西他滨(0.1 μM)处理 6 天后,OCI-Ly8 细胞中的甲基化逐渐降低,miRNA-363-3p 水平显著升高。此外,与 OCI-Ly3 相比,MYC 的异位表达并没有明显增加 OCI-Ly8 中 miRNA-363-3p 的水平(图 2E)。总之,这些结果证实了 miRNA-363-3p 受 MYC 以外的 DNA 甲基化的转录调控。
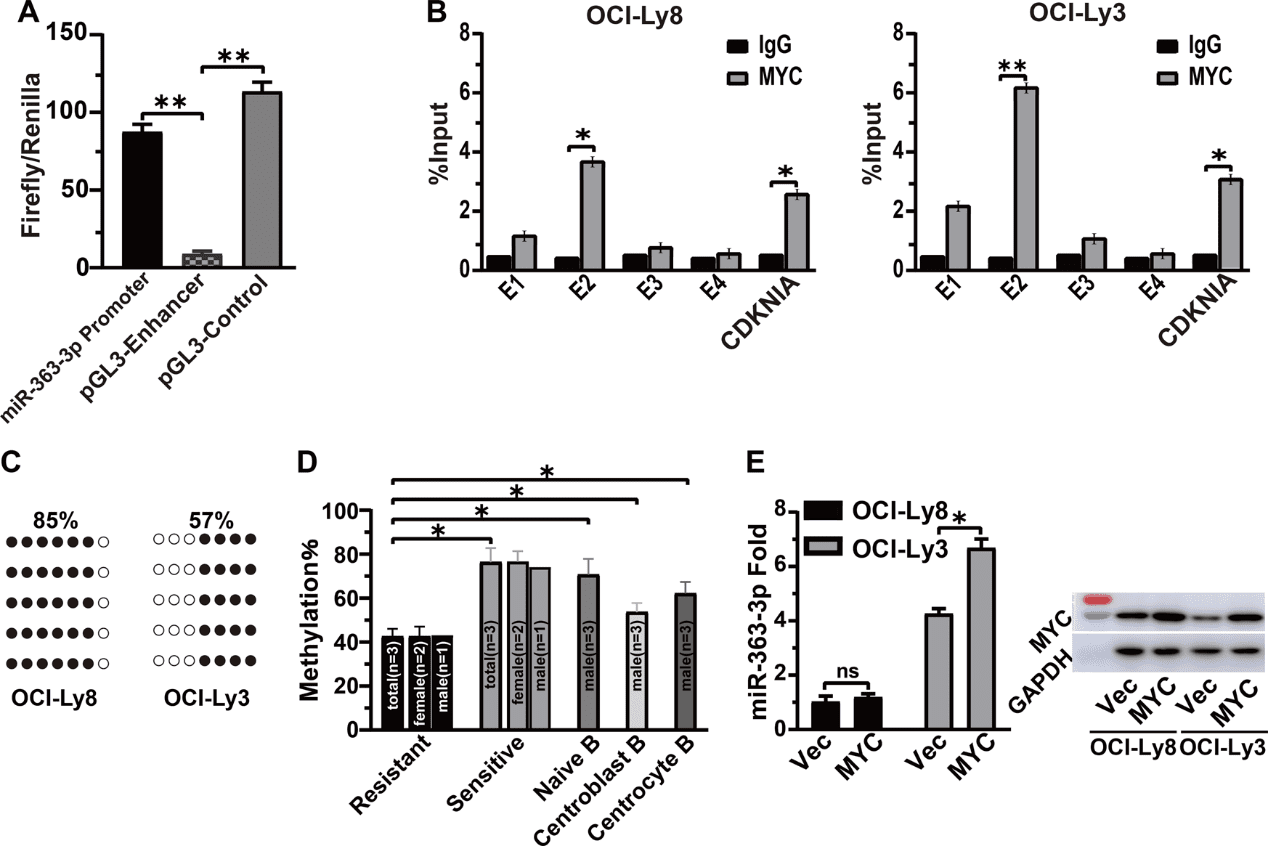 图2. miRNA-363-3p 的转录调控。
图2. miRNA-363-3p 的转录调控。
对来自 47 名 DLBCL 患者冷冻样本的 GEP 数据进行了无监督的分层聚类,将患者分为两个不同的亚组。一组(n = 16)的所有患者都是耐药的,而另一组的大多数(29/31)病例是敏感的。总共有 78 个上调基因(倍数变化 > 2 和 p < 0.01)和 135 个下调基因(倍数变化 < 0.5 和 p < 0.01)被富集为与抗性相关。此外,预测了 278 个 miRNA-363-3p 的潜在靶转录物。当研究人员将下调的基因与 miRNA-363-3p 的候选靶基因重叠时,发现了 DUSP10、FOXG1 和 CACNA1C。为了测试 miRNA-363-3p 对这些基因的直接调控,将候选基因的 3′UTR 与推定的或突变的结合点克隆到荧光素酶报告质粒中。结果显示,miRNA-363-3p 模拟物显著抑制了含有 DUSP10 和 CACNA1C 基因的 3'UTR 质粒的荧光素酶活性,但没有抑制 FOXG1 基因的 3'UTR(图 3A)。Western blot 分析证实,DUSP10 蛋白在 miRNA-363-3p 异位细胞系 OCI-Ly8 中减少,在 miRNA-363-3p 敲除细胞系 OCI-ly3 中增加(图 3B)。
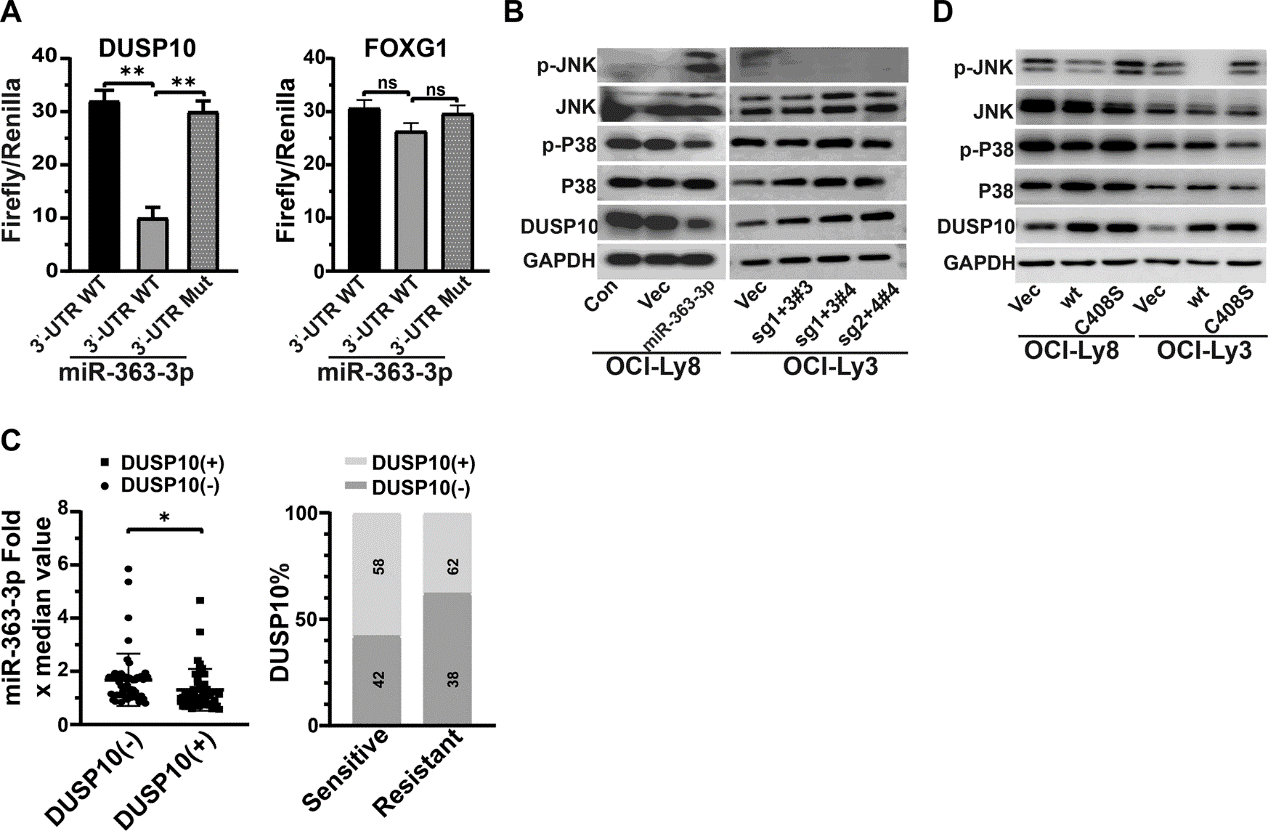 图3. DUSP10 在 miRNA-363-3p 介导的化疗耐药性中的作用。
图3. DUSP10 在 miRNA-363-3p 介导的化疗耐药性中的作用。
在 106 名 DLBCL 患者中验证了 DUSP10 表达与 miRNA-363-3p 和化疗耐药性的负相关关系(图 3C)。在另一个更大的 DLBCL 队列(n = 221)中,具有 DUSP10 基因低表达的患者也显示出不利的 PFS 和总生存期(Overall survival, OS)。这些数据证实 DUSP10 是 miRNA-363-3p 的一个靶基因,并促成了 DLBCL 中与 miRNA-363-3p 相关的耐药性。JNK 和 p38 受到 DUSP10 介导的去磷酸化的负调节,涉及丝裂原活化蛋白激酶(MAPK)信号通路。此外,还发现磷酸化 JNK 的水平在 miRNA-363-3p 异位细胞系 OCI-Ly8 中增强,在 miRNA-363-3p 敲除细胞系 OCI-Ly3 中降低(图 3B)。磷酸酶死亡的 DUSP10(C408S)进一步验证了 miRNA-363-3p/DUSP10/JNK 轴在 DLBCL 细胞中的功能(图 3D)。
对 78 个耐药性相关的上调基因进行了 David 通路分析,发现前 10 种通路;DNA 损伤修复途径主要被富集,包括 HR 的负调控和 DNA 修复(图 4A),其在 miRNA-363-3p 异位细胞系 OCI-Ly8 和 miRNA-363-3p 敲除细胞系 OCI-Ly3 细胞中得到进一步验证。多柔比星诱导的 miRNA-363-3p 异位 OCI-Ly8 细胞中的 γH2AX 水平在多柔比星(25 ng/ml)处理 2 h 后的 0、2、6、12 和 24 h 逐渐降低,在 OCI-Ly8 对照细胞中没有减少,γH2AX 是已知的双链断裂(double-strand break, DSB)共识生物标志物。相反,miRNA-363-3p 敲除导致 OCI-Ly3 细胞中多柔比星诱导的 γH2AX 水平增加(图 4B)。然而,并未发现 TP53 和凋亡相关途径的基因在这些 miRNA-363-3p 高表达的患者中富集。miRNA-363-3p 表达的变化不影响 TP53 和 BCL2 蛋白的水平。与预期一致,ATF2 和磷酸化 ATF2 的水平与 miRNA-363-3p 表达呈正相关(图 4C),这是 JNK 的一个共识靶标。
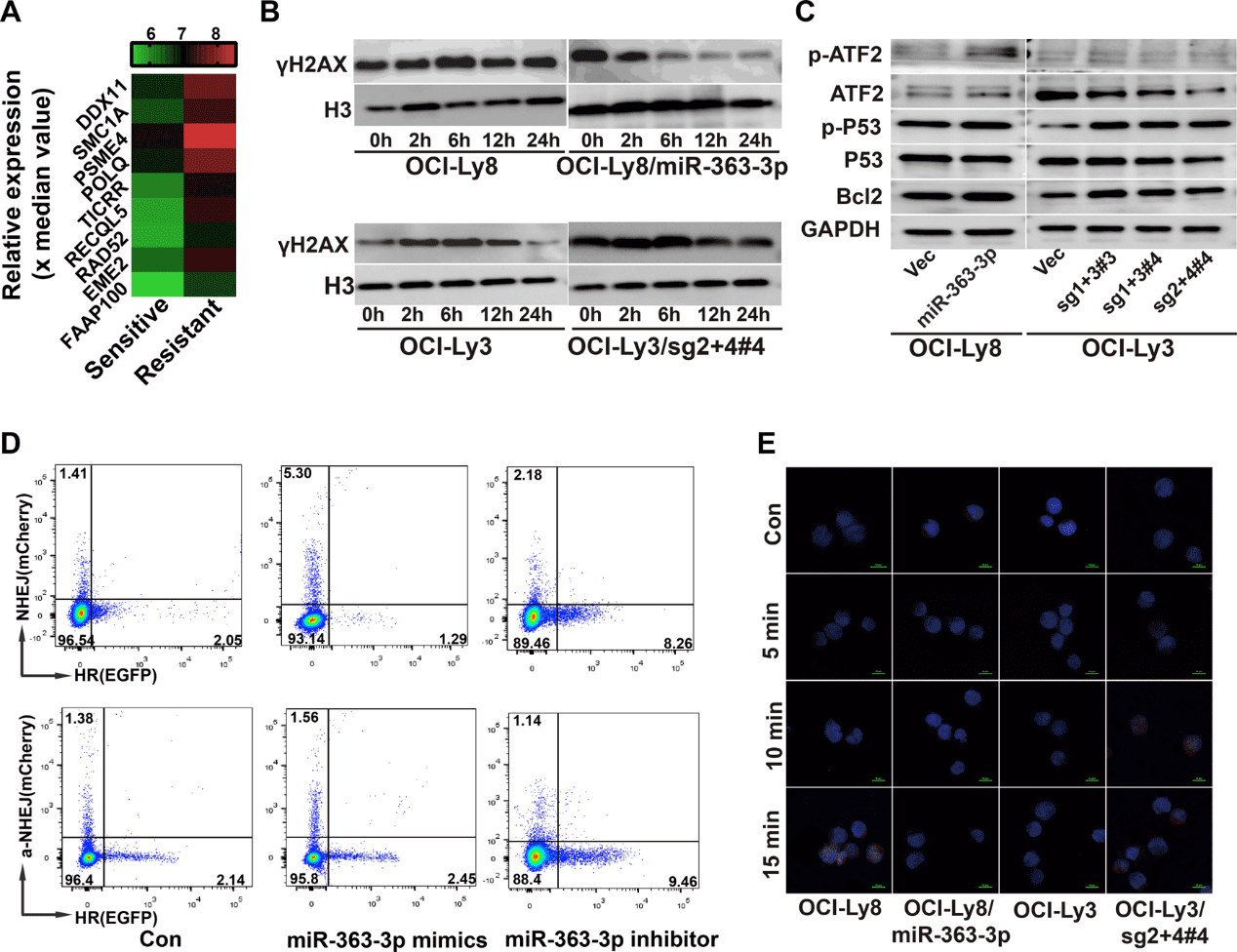 图4. miRNA-363-3p/DUSP10/JNK 轴相关化学耐药性中的异常 DNA 损伤修复。
图4. miRNA-363-3p/DUSP10/JNK 轴相关化学耐药性中的异常 DNA 损伤修复。
DSB 主要由 HR(Homologous recombination)和 NHEJ(Non-homologous end-joining)两种机制修复,偶尔也由容易出错的替代性 NHEJ 修复。使用报告质粒系统,在 293 T 细胞中验证了 miRNA-363-3p 模拟物可显著降低 HR 活性,增加 NHEJ 和替代 NHEJ 活性;miRNA-363-3p 抑制剂则表现出相反的效果(图 4D)。RAD51 是 HR 修复的一个核心分子。免疫荧光测定发现多柔比星诱导的 RAD51 病灶因 miRNA-363-3p 的异位表达而明显减少,但因 miRNA-363-3p 的敲除而增加(图 4E)。最终证实了 miRNA-363-3p 诱导 HR 抑制。
PARP1 抑制剂已被证明对 HR 缺陷型癌细胞具有显著疗效。因此,评估了 pan-JNK 抑制剂 SP600125 和 PARP1 抑制剂 BGB-290 在 DLBCL 细胞中克服与 miRNA-363-3p/DUSP10/JNK 轴相关的化学耐药性的潜力。结果显示,它们在 miRNA-363-3p 高表达的 OCI-Ly3 细胞中明显增强了多柔比星(25 ng/ml)诱导的 γH2AX 水平(图 5A)。此外,在 miRNA-363-3p 高表达的细胞系中观察到多柔比星和 SP600125 或 BGB-290 之间存在明显的协同作用,但在低表达的细胞系中则没有观察到(图 5B)。它们的疗效在携带 OCI-Ly3 细胞的异种移植小鼠模型中得到进一步证实,SP600125 或 BGB-290 的添加显著增加了多柔比星诱导的肿瘤消退和生存率(图 5C)。总之,该研究证明了通过抑制 DLBCL 细胞中的 JNK 和 PARP1 可以逆转 miRNA-363-3p 相关的化疗耐药性,内在机制如图 5D 所示。
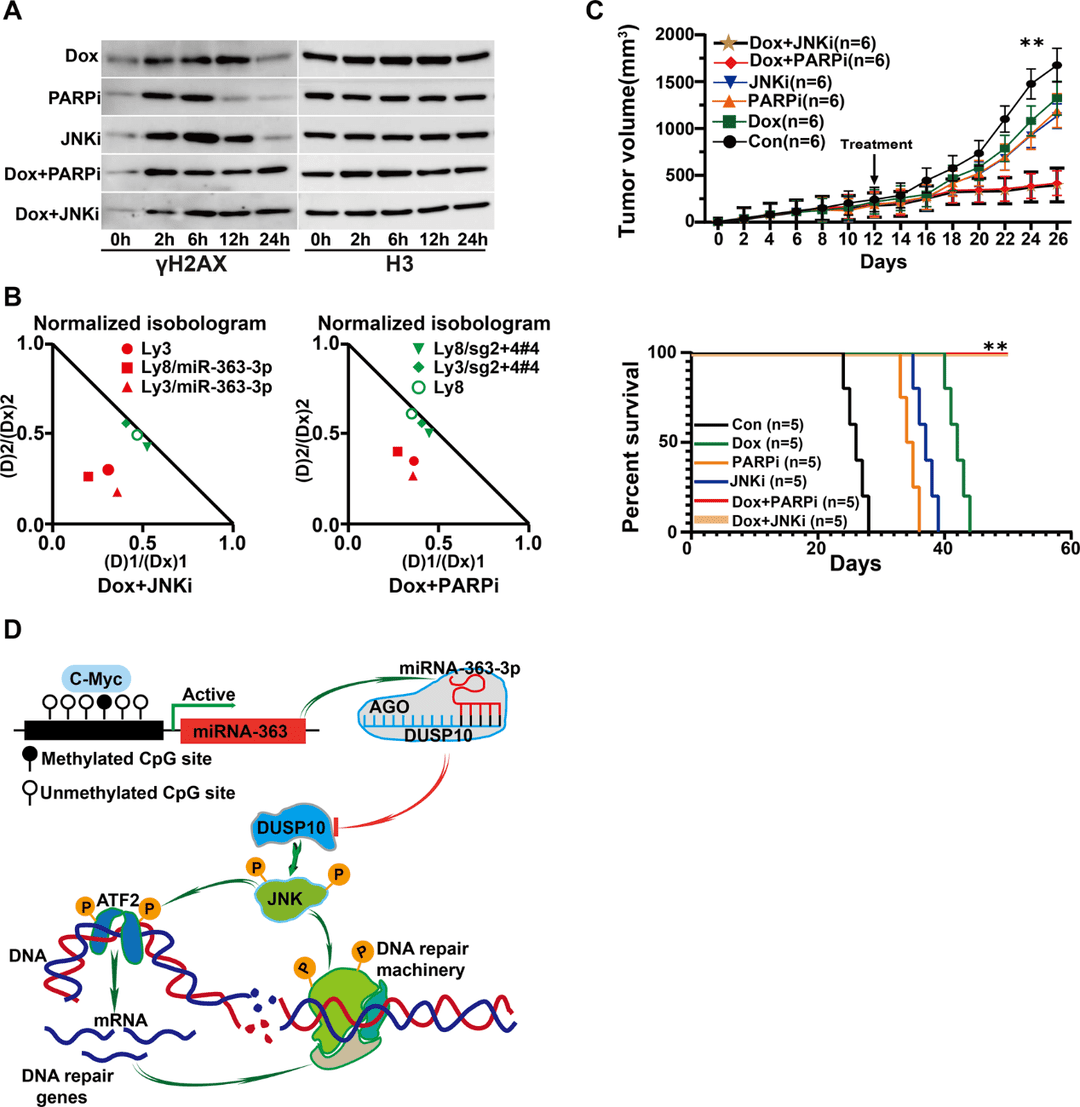 图5. JNK 和 PARP1 抑制剂对多柔比星耐药的影响。
图5. JNK 和 PARP1 抑制剂对多柔比星耐药的影响。
该研究确定了 miRNA-363-3p 与化疗耐药性之间的独特关联。证实了除 MYC 以外的 DNA 甲基化对 miRNA-363-3p 的特异性转录调控,这与 miRNA-17-92 簇和来自 miRNA-106a-363 的 miRNA-18b、miRNA-20b 和 miRNA-106a 等由 MYC 上调的集群有所差别。已发现 miRNA-363-3p 的失调与某些实体瘤的肿瘤发生、增殖和耐药性有关。研究表明,敲除 miRNA-363-3p 具有抗肿瘤活性。miRNA 传递系统的发展将推动 miRNA 疗法的临床转化。此外,DSB 是化疗药物引起的最为危险的 DNA 损伤,内在的 DSB 修复能力决定了肿瘤细胞的命运。该研究证明了 miRNA-363-3p 相关的化学耐药性可归因于 DLBCL 中增强的 DSB 修复功能。总之,该研究创造性地提出了一种新的基于蒽环类药物的化学耐药性机制,涉及 miRNA-363-3p/DUSP10/JNK 介导的 DSB 修复增强。探究针对这种耐药机制的新方法为克服化学耐药性提供了潜在的选择。
原文链接:https://www.nature.com/articles/s41375-022-01565-6
安必奇生物致力于为国内外客户提供 MicroRNA Agomir/Antagomir 相关综合服务。全面的服务涵盖 miRNA Agomir/ Antagomir 的设计,合成,筛选,递送以及应用开发等各个环节。为您的科学研究提供更加高效的解决方案。欢迎免费咨询!
24小时服务在线

