B 细胞淋巴瘤,包括慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)和弥漫性大 B 细胞淋巴瘤(diffuse large B-cell lymphoma, DLBCL),是非霍奇金淋巴瘤(non-Hodgkin lymphoma, NHL)中最常见的亚型,其特点是 B 细胞受体(B-cell receptor, BCR)信号的慢性激活。布鲁顿酪氨酸激酶(Bruton’s tyrosine kinase, BTK)抑制剂(BTKi)已在新诊断和治疗复发/难治性 CLL 和许多 NHL 亚型患者中显示出令人印象深刻的临床活性,但随着时间的推移,许多接受 Ibrutinib(FDA 批准的口服 BTK 抑制剂)治疗的患者会复发或产生耐药性。
Acalabrutinib 是 FDA 批准的第二代、高选择性、强效、共价 BTK 抑制剂,具有最小的脱靶效应,在初治、复发/难治性 CLL 患者中显示出良好的耐受性和疗效。与 Ibrutinib 相比,具有相似的临床活性,但安全性有所提高。之前的研究表明对 Ibrutinib 的获得性耐药与肿瘤抑制蛋白 PTEN 的下调和 PI3K/AKT 通路的激活有关。然而,PTEN 如何介导 B 细胞恶性肿瘤中对 BTK 抑制的抗性尚不清楚。
2021 年 11 月 8 日,美国俄亥俄州克利夫兰勒纳研究所的研究人员在 Cell Death & Disease 杂志上发表了题为“协同 miRNA 依赖性 PTEN 调节驱动 B 细胞淋巴恶性肿瘤对 BTK 抑制的抗性”的研究论文。该研究展示了 BTKi ibrutinib 和 acalabrutinib 下调位于 14q32 miRNA 簇区域的 miRNA,包括 miR-494、miR-495 和 miR-543。BTKi 抗性 CLL 和 DLBCL 细胞显著过表达 miR-494、miR-495、miR-543,并降低 PTEN 表达,表明 PI3K/AKT/mTOR 通路在获得性 BTKi 抗性中得到进一步调节。此外,miR-494 模拟物的过表达降低了 PTEN mRNA 和蛋白质水平,进一步表明 PTEN/AKT/mTOR 对细胞凋亡的调节。相反,在 BTKi 抗性细胞中过表达 miR-494 抑制剂可恢复 PTEN mRNA 和蛋白质水平,从而使细胞对 BTKi 诱导的细胞凋亡敏感。因此,靶向 14q32 簇 miRNA 可能通过调节 PTEN/AKT/mTOR 信号轴对获得性 BTK 耐药患者具有治疗价值。

通过体外培养亲本细胞系时用逐渐增加浓度的 acalabrutinib 或 ibrutinib 处理产生 Aca-R ABC-DLBCL(TMD8)、IB-R ABC-DLBCL(RIVA,TMD8)和CLL(MEC-1)细胞系。细胞活力分析显示,在 acalabrutinib 处理 24 h 后,TMD8 的细胞死亡增加了约 40%(图1a),但Aca-R(acalabrutinib-resistant)-衍生细胞没有。同样,MTS 检测显示对增加浓度的 acalabrutinib 的敏感性很高。免疫印迹分析表明,与亲本细胞相比,抗性细胞(TMD8-Aca-R)中 PTEN 的水平较低(图1b)。此外,与亲本细胞相比,TMD8-Aca-R 中 pAKT(AKTSer473)的水平上调。而 pBTK(BTKY223)的水平降低了,表明长期的 acalabrutinib 处理阻断了 Aca-R 细胞中 BTK 的激活。值得注意的是,qRT-PCR 分析发现 pten mRNA 水平降低了约 4 倍,表明 PTEN 水平降低可归因于 pten mRNA 水平的降低(图1c)。总之,这些发现表明 PTEN/AKT 轴在介导 Aca-R 中的重要性。
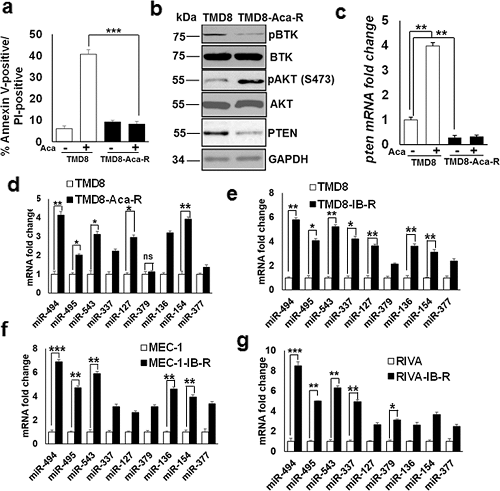 图1. BTK 抑制下调 CLL 和 DLBCL 细胞中 14q32 簇区域的 miRNAs。
图1. BTK 抑制下调 CLL 和 DLBCL 细胞中 14q32 簇区域的 miRNAs。
为研究对 BTK 抑制的耐药机制,研究人员检查了位于 14q32 簇的 miRNA 的表达模式,在之前发现这些 miRNA 与 CLL 和 DLBCL 中对 BCL-xL 抑制的耐药性有关。通过 qRT-PCR 分析检查位于 14q32 簇中的 9 种 miRNA 的表达模式,表明它们在 Aca-R、IB-R DLBCL 和 CLL 细胞系中的表达增加。在这些 miRNA 中,miR-494 和 miR-543 在 TMD8-Aca-R 细胞中的表达分别增加了~ 3 倍和 2 倍(图1d)。在 TMD-IB-R 中,miR-494、miR-495 和 miR-543 的表达分别增加了 ~ 4.8 倍、~ 3 倍和 ~ 2 倍(图1e),在 MEC-1-IB-R 中分别为 ~ 6 倍、4 倍和 5 倍(图1f),在 RIVA-IB-R 细胞中分别为 ~ 7.5 倍、4 倍和 5 倍(图1g)。总之,这些发现表明 14q32 簇 miRNA 的异常表达与 BTK 抑制的耐药性有关。
由于长期暴露于 acalabrutinib 或 ibrutinib,BTKi 耐药导致 14q32 簇 miRNAs 的表达增加,PTEN 水平降低。接下来研究了急性 acalabrutinib 和 ibrutinib 处理对 BTki 敏感的 ABC-DLBCL 和CLL 细胞中 14q32 簇 miRNAs 的影响。用 acalabrutinib 处理的亲本 TMD8 细胞显示出 14q32 簇 miRNAs 的下调,miR-494(~50%)、miR-495(~40%)和 miR-543(~30%)水平降低(图2a)。Ibrutinib 在 TMD8 细胞中对 BTK 的抑制导致 miR-494 和 miR-543 的表达降低约 90%,miR-495 的表达降低约 80%(图2b)。类似地,在 MEC-1(图2c)和 RIVA 细胞(图2d)中也观察到 miR-494、miR-495 和 miR-543 的表达降低了约 90%。总之,这些发现表明 14q32 簇 miRNA 的异常表达在介导 BTKi 耐药性方面具有潜在作用。
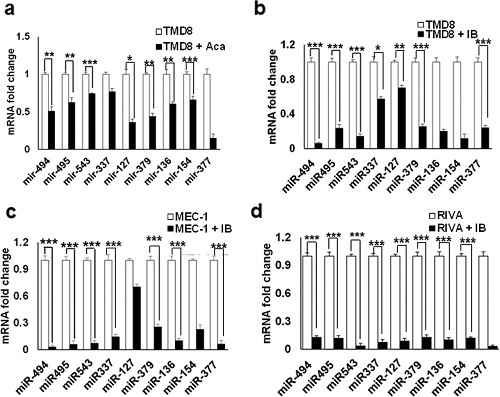 图2. CLL 和 DLBCL 中 BTK 抑制后 14q32 簇 miRNA 的表达降低。
图2. CLL 和 DLBCL 中 BTK 抑制后 14q32 簇 miRNA 的表达降低。
鉴于体外 Aca-R 和 IB-R CLL 和 DLBCL 细胞中 14q32 簇 miRNAs 和 PTEN 表达的差异,接下来测试了 miRNA 表达是否也可能在患者来源的原发性 CLL 细胞中发生改变,以响应 BTK 抑制或标准的临床治疗。qRT-PCR 分析了临床上 ibrutinib 治疗前后的三对 CLL 患者样本,结果显示,临床治疗后 #CLL3(ibrutinib 敏感型)中 miR-494、miR-495 和 miR-543 的水平降低,与 #CLL1(ibrutinib 耐药型)和 #CLL2(部分缓解型)相反(图3a)。此外,ibrutinib 对 CLL 患者样本的体外处理显示,在治疗复发患者与初治患者中,miR-494、miR-495 和 miR-543 的表达增加(图3b)。而体外 acalabrutinib 处理初治(#CLL4, #CLL5)与治疗复发(#CLL6, #CLL7)的 CLL 患者,发现 #CLL4(2.9 倍)和 #CLL5(3.2 倍)的 pten mRNA 水平显著增加,与 #CLL6 和 #CLL7 相反(图3d)。综上,这些结果表明 14q32 簇 miRNAs 的异常表达在介导治疗抗性中发挥作用。
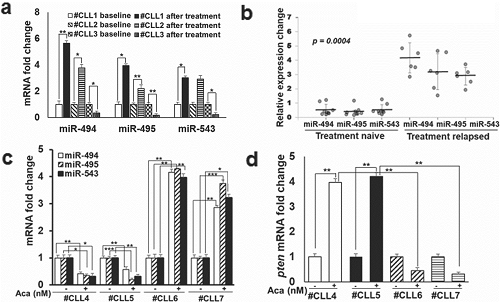 图3.在患者来源的原代 CLL 细胞中,BTK 抑制后 14q32 簇 miRNA 的表达降低,而 PTEN 的表达增加。
图3.在患者来源的原代 CLL 细胞中,BTK 抑制后 14q32 簇 miRNA 的表达降低,而 PTEN 的表达增加。
之前的研究表明,ibrutinib 治疗可调节 CLL 和 DLBCL 细胞中 PTEN 的转录激活。使用靶点预测软件来确定具有假定 PTEN 靶点的 miRNA,研究人员发现了 6 个位于 14q32 簇的 miRNA,其中 miR-494 的得分最高。对转染了 miR-494 抑制剂的 TMD8-Aca-R 细胞进行 qRT-PCR 和免疫印迹分析,发现 pten mRNA(上图)和蛋白(下图)的表达水平明显增加(图4a)。在 TMD8-IB-R(图4c)、MEC-1-IB-R 和 RIVA-IB-R 细胞中也得到类似的结果。为了证实 miR-494 直接参与 PTEN 的调控,加入 miR-494 模拟物导致亲代 TMD8(图4b)、MEC-1 和 RIVA 细胞中内源性 PTEN mRNA(上图)和蛋白(下图)表达水平均大幅下降。这些结果表明 PTEN 受 miR-494 的转录后水平调节,并且可能是直接靶标。
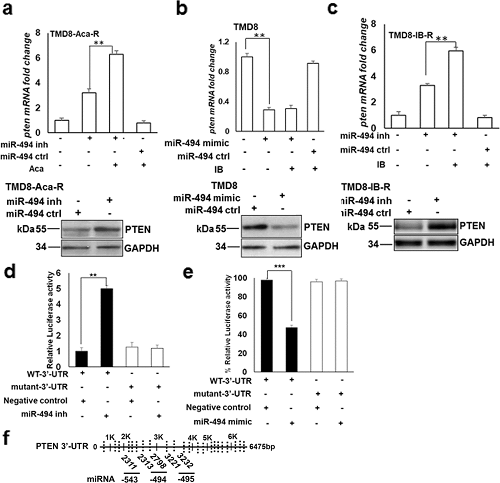 图4. PTEN 是 CLL 和 DLBCL 中 miR-494 的直接靶标。
图4. PTEN 是 CLL 和 DLBCL 中 miR-494 的直接靶标。
为了确定 miR-494 的潜在靶标,研究人员使用 mirDB 对 14q32 miRNA 簇区域和 PTEN 3'-UTR 序列进行了预测分析。该分析在 PTEN mRNA 的 3'-UTR 的 2313 和 2798 处鉴定出了两个保守的互补序列,miR-494 可能与之配对(图4f)。为了研究 PTEN 是否是 miR-494 的直接靶标,将含有野生型(WT)PTEN 3'-UTR 或 miR-494 结合位点突变体 PTEN 3'-UTR 的荧光素酶报告基因与 miR-494 模拟物(图4e)或抑制剂(图4d)一起转染 TMD8 和 TMD8-IB-R 细胞,48 h 后测量荧光素酶活性。与阴性对照相比,TMD8 细胞中的异位 miR-494 模拟物的表达使 WT-3'-UTR 相关荧光素酶活性下调约 50%,而转染 mutant 3′-UTR 荧光素酶报告基因,miR-494 模拟物无法抑制荧光素酶活性(图4e)。相比之下,与阴性对照相比,在 TMD8-IB-R 细胞中用 miR-494 抑制剂转染完全逆转了荧光素酶活性,导致 WT-3'-UTR 相关荧光素酶活性增加 4 倍(图4d)。总之,这些结果表明 miR-494 与 PTEN 3'-UTR 中预测的和先前报道的靶点直接结合。
接下来研究了是否可以结合 miR-494 或 miR-495,通过 AKT 抑制使得这些细胞进一步对凋亡敏感。之前的研究已经表明 IB-R 细胞中的 AKT 激活升高,而 PTEN 显著下调,从而使 IB-R 细胞对通过 AKT 抑制诱导的细胞凋亡更敏感。对 AKT 的药理学抑制显示 TMD8-Aca-R 细胞的凋亡分别增加了 ~ 20% 和 ~ 19%,分别是通过 miR-494(图5a)或 miR-495(图5b)抑制 AKT,在 TMD8-IB-R 细胞中结果类似(图5c,5d)。这些结果共同表明 BTKi-R 细胞依赖 miR-494 或 miR-495 依赖性的 PTEN/AKT 调节,抑制 AKT 的磷酸化/激活会增加 miR-494 和 miR-495 抑制诱导的 BTK-R 细胞的凋亡。
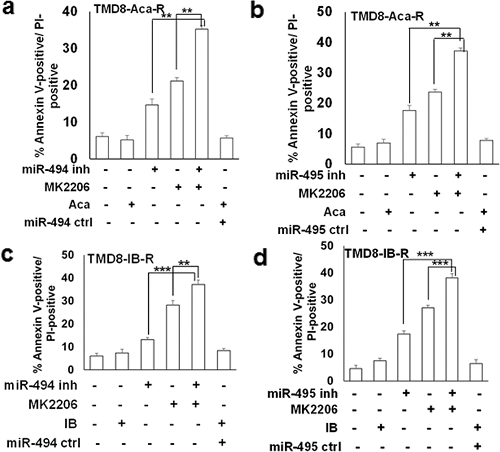 图5. 在 BTK 抑制剂耐药的 CLL 和 DLBCL 中,miRNA 抑制增强了 AKT 诱导的细胞凋亡。
图5. 在 BTK 抑制剂耐药的 CLL 和 DLBCL 中,miRNA 抑制增强了 AKT 诱导的细胞凋亡。
miR-494 抑制剂抑制了 AKT 和 mTOR 的活性,如 pAKT Ser473 的减少和 mTORC1、p70S6 Thr389 激酶和 p4EBP1 下游靶标磷酸化的抑制所示。值得注意的是,通过抑制 miR-494,TMD8-IB-R 细胞中 PTEN 蛋白水平的表达增加。然而,AKT 和 p70S6 激酶水平没有显著变化(图6a)。这些发现表明,TMD8-IB-R 细胞中响应于 miR-494 抑制的 PTEN 蛋白表达增加与 AKT 和 mTOR 活性降低有关。此外,miR-494 抑制导致 caspase 3 cleavage 的表达增加(图6a),抑制 miR-495 获得了类似的结果(图6b)。接下来,研究人员检查了这些 miRNA 是否可以通过调节 PTEN 表达协同影响细胞活力。在 TMD8-IB-R(图6c)和 MEC-1-IB-R(图6d)细胞中的细胞活力分析表明,与单独转染 miR-494 或 miR-495 抑制剂相比,在转染了 miR-494 和 miR-495 抑制剂组合的细胞中,细胞凋亡增加了约 20%,表明这些 miRNA 抑制剂对细胞存活具有协同作用。
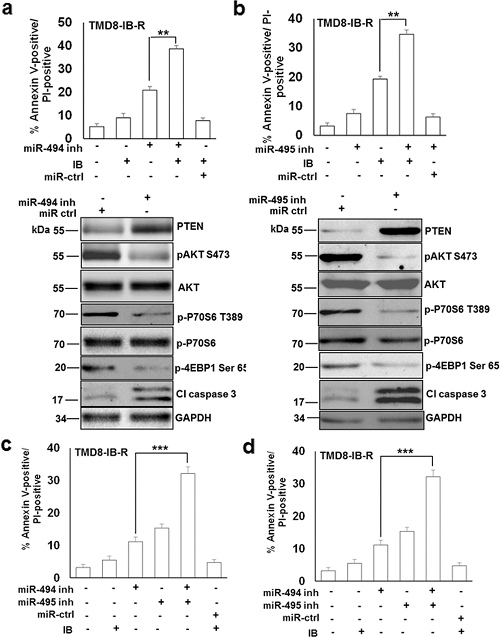 图6. miR-494 和 miR-495 的协同抑制通过 AKT/mTOR 信号传导增强细胞存活。
图6. miR-494 和 miR-495 的协同抑制通过 AKT/mTOR 信号传导增强细胞存活。
尽管 B 细胞淋巴恶性肿瘤存在广泛的异质性,但越来越多的证据支持 miRNAs 表达失调与各种癌症(包括 CLL 和 DLBCL)治疗的耐药性之间存在关联。最近的发现表征了通过 PTEN 下调阻碍 ibrutinib 诱导的细胞凋亡的获得性耐药性的发展。该研究的发现为 BTK 抑制剂耐药机制提供了新的分子见解,并支持 Aca-R 和 IB-R 细胞中 14q32 簇 miRNAs 的异常表达与 anti-miR-494 或 miR-495 上调 PTEN 的能力之间的联系,以克服耐药性并通过减弱 AKT 的激活诱导细胞凋亡。因此,14q32 miRNA 簇/PTEN/AKT/mTOR 轴成为 CLL 和 DLBCL 中获得性 BTKi-R 的决定因素,将 miRNA 和 AKT 抑制组合作为一种合理的组合策略,使 BTKi-R 细胞对凋亡敏感,对于耐药性肿瘤的疗法开发意义重大。
原文链接:https://www.nature.com/articles/s41419-021-04353-9
安必奇生物致力于为国内外客户提供 MicroRNA Agomir/Antagomir 相关综合服务。全面的服务涵盖 miRNA Agomir/ Antagomir 的设计,合成,筛选,递送以及应用开发等各个环节。为您的科学研究提供更加高效的解决方案。欢迎免费咨询!
24小时服务在线

